I-PINE requires protein sequence file and peak lists from solution-state NMR experiments. Optional inputs to enhance the overall quality are also accepted. More detailed description for each file is written below.
Sequence
A sequence file in text file format is required. A RTF (Rich Text Format; .rtf), an ODT (OpenDocument Text; .odt), and a DOCX (Office Open XML; .docx) are automatically converted to the text file. I-PINE accepts either one-letter and three-letter. All the letters in the file must be in capital letters. Please note that appropriate file extensions in file names are mandatory for non-ASCII text files.
Peak Lists
Peak lists in (NMRFAM/UCSF)-Sparky, XEASY, NMRView, NMRDraw, and I-PINE simple formats are accepted as inputs. At least, one root experiment (2D N-HSQC or 3D HNCO), and one 3D experiments focused on CA is required (e.g. HNCACB). However, more experiments provide more robustnesss and higher chances of successful assignments because of the Bayesian probabilistic framework of the I-PINE. Profiles for supported experiments are described here: [click]
Prot file (optional)
An empty .prot file is necessary to be submitted if a user wants to get assignments in Xeasy .prot format.
Pre-assignment (optional)
If a user wants to use chemical shifts of some residues already assigned, a pre-assignment file can be used. The file is NMR-STAR-like format and I-PINE will prioritize assignments in this file first when finding possible assignments for resonances. However, if given assignments do not make sense at all considering inputs, the I-PINE will disregard and try to assign others. Therefore, it is highly recommended to use the PINE-SPARKY.2 plug-in to use correctly aligned assignments and peak lists. This feature is particularly useful if the protein is too difficult for I-PINE to assign all the residues at first run, because it is possible for the user to increase the level of completeness gradually by iterative employment of automation with partial assignments.
On the other hand, the pre-assignment file containing almost complete chemical shifts can be used as an only input other than sequence information when a user wants to conduct chemical shift based analyses that I-PINE offers. I-PINE generates synthetic peak lists from the pre-assignment file (N15-HSQC, HNCO, HN(CA)CO, HN(CO)CA, HNCA, HN(CO)CACB, HNCACB, C13-HSQC, HA(CO)NH, HNHA, HBHA(CO)NH, HBHANH, C(CO)NH, H(CCO)NH, TOCSY-HSQC, CCH-TOCSY, HCCH-TOCSY) and conducts automated analysis. By this way, a user does not have to go through different formats and installations and executions of different software packages but only to run I-PINE and see all the results in a provided I-PINE web results page.
Selective labeling (optional)
Basically, the selective labeling strategy is used to make spectra less crowded and overlapped for easy interpretation of data. I-PINE makes use of this information to give amino acid type preferences to the 2D N-HSQC peaks. For instance, a user knows Isoleucine (I), Leucine (L) and Valine (V) peaks in the 2D N-HSQC, a user can prepare a supplement file describing which peaks should be ILVs. Another application is to use Serine (S) and Threonine (T) information. As Serines and Threonines have distinctively different characteristics than other amino acid types (CB (ppm) > CA (ppm)), they can be easily pointed from 3D experiments (e.g. HNCACB). This kind of information can be also used to help the I-PINE to reduce the number of choices. In the NMRFAM-SPARKY, using this feature is very easy because the PINE-SPARKY.2 automatically generates the selective labeling input file using user labels.
Atomic coordinate (optional)
Since the I-PINE accepts 3D N15-NOESY, it is also possible to use atomic coordinates (PDB or PDBx/mmcif file) to improve the assignments. The I-PINE looks given NOESY data to rebalance probabilities and connectivities. Residue numbers in the PDB are recommended to match with the other residue numbering in the I-PINE, however, the I-PINE will try to adjust automatically. The given 3D N15-NOESY and C13-NOESY data are passed to PONDEROSA-C/S to assign NOE cross peaks. A user will receive separate emails from PONDEROSA web server regarding assigned NOESY data and 3D structures when an email address is provided. Otherwise, results will be automatically included in their results associated with the same job identification number accessible by the URL provided right after the submission.
Spin Systems (replacement for peak lists)
I-PINE generates spin system matrix using peak lists and orders spin systems based on connectivity matrix between them before going into the probabilistic assignment procedure. However, this step can be skipped by providing a spin system file instead.
Run times vary depending on clearity of the inputs and size of the proteins. Only a few minutes is needed, however, it can go over a couple of hours as well if the protein is pretty larger. Results will be sent to the user email when an email address is provided, and please contact us if you do not get anything in a few hours (check spam inbox, too). However, user information is only optional when a user chooses to receive a result email, and all the results are accessble from a job identification nubmber provieded right after the submission.
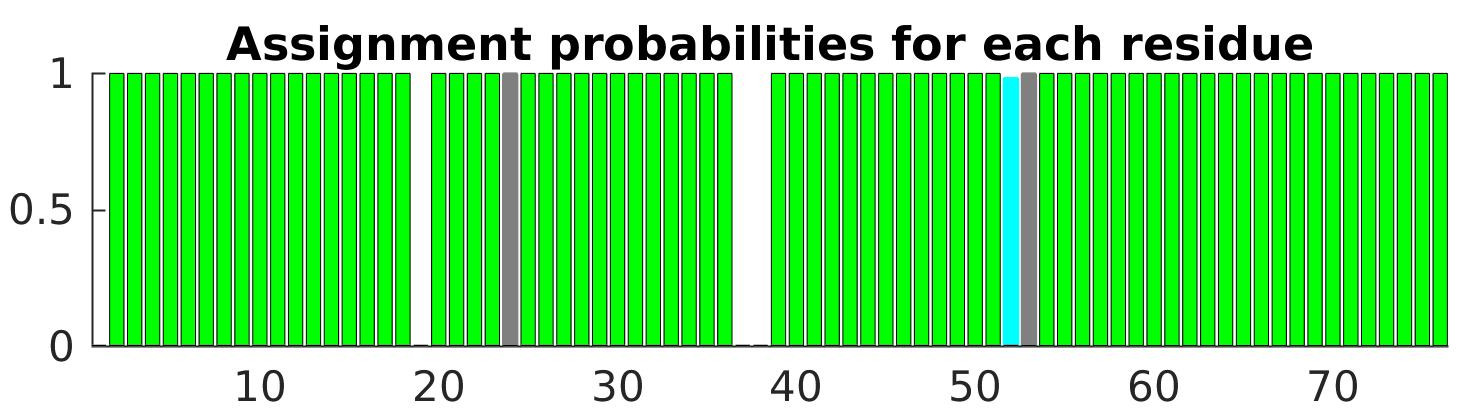
Probability bar graph
Probability bar graph (protein.jpg) is a 2D bar plot graph visualizing assignment probabilities for each residue with different color codes; green bars (> 0.99); cyan bars (0.85 - 0.99); yellow bars (0.50 - 0.85); red bars (< 0.50); gray bars (no assignment). A NDPPLOT format (ndpplot_PINE.ini) is also provided. The NDPPLOT is an interactive 2D plotting tool in the NMRFAM-SPARKY software package.
2D N-HSQC plot
 2D N-HSQC plot (hsqc_plot.svg) is a plot showing synthesized N-HSQC peaks based on I-PINE chemical shift assignments with color codes. The colors are similar to the probability bar graph but just darker for better visual recognition (dark green, blue, mustard, red). Because the plot is in SVG (scalable vector graphics) format, it does not pixellize and also a user can use "find" and "zoom" tool in the web browser.
2D N-HSQC plot (hsqc_plot.svg) is a plot showing synthesized N-HSQC peaks based on I-PINE chemical shift assignments with color codes. The colors are similar to the probability bar graph but just darker for better visual recognition (dark green, blue, mustard, red). Because the plot is in SVG (scalable vector graphics) format, it does not pixellize and also a user can use "find" and "zoom" tool in the web browser.
Chemical shifts
Assigned chemical shifts are provided in five different file formats. I-PINE probabilistic backbone assignments (protein_backbone_assignment.txt) and sidechain assignments (sidechain_table.txt), BMRB NMR-STAR 2.1 (protein_nmrstar21.str), 3.1 (protein_nmrstar31.str) and 3.2 (protein_nmrstar32.str) and SPARKY resonance list (sparkyresonances.list).
Assigned peak lists
I-PINE puts out assigned peak lists using user submitted peak lists and probabilistic assignments. Basically, SPARKY peak lists with an assignment label, chemical shift of each dimension and a probability are provided. Assigned XEASY peak lists are provided when a user uploaded the empty prot file. Also, BMRB NMR-STAR 3.2 includes assigned peak lists.
Secondary structures
 I-PINE provides PECAN secondary structure predictions (Eghbalnia et al. 2005) based on backbone chemical shifts assigned by I-PINE algorithm. TALOS-N results from I-PINE backbone chemical shifts also include secondary structure predictions (see below). A 2D plot image (pecan_fig.jpg, plot_PECAN.svg), an NDP-PLOT file (ndpplot_PECAN.ini) and a probabilistic text file (temp_PECAN.out) are provided. Helices are illustrated in green and strands are in blue. GetSBY (Get Secondary Structure By PACSY; Lee & Lee 2009 KR Patent 1,008,899,400,000; Shin, Lee & Lee 2008) also provides probabilistic secondary structure prediction results (report_GETSBY.txt, ndpplot_GETSBY.ini, plot_GETSBY.svg). Additionally, we provide secondary chemical shift analysis on CA and CB by calculating (CAobserved-CAcoil) - (CBobserved-CBcoil) for each residue (secshift.txt, ndpplot_SSHIFT.ini, plot_SSHIFT.svg). This CSI (Chemical Shift Index) approach has been known to predict accurate protein secondary structures as high as 92% (Wishart & Sykes. 1994).
I-PINE provides PECAN secondary structure predictions (Eghbalnia et al. 2005) based on backbone chemical shifts assigned by I-PINE algorithm. TALOS-N results from I-PINE backbone chemical shifts also include secondary structure predictions (see below). A 2D plot image (pecan_fig.jpg, plot_PECAN.svg), an NDP-PLOT file (ndpplot_PECAN.ini) and a probabilistic text file (temp_PECAN.out) are provided. Helices are illustrated in green and strands are in blue. GetSBY (Get Secondary Structure By PACSY; Lee & Lee 2009 KR Patent 1,008,899,400,000; Shin, Lee & Lee 2008) also provides probabilistic secondary structure prediction results (report_GETSBY.txt, ndpplot_GETSBY.ini, plot_GETSBY.svg). Additionally, we provide secondary chemical shift analysis on CA and CB by calculating (CAobserved-CAcoil) - (CBobserved-CBcoil) for each residue (secshift.txt, ndpplot_SSHIFT.ini, plot_SSHIFT.svg). This CSI (Chemical Shift Index) approach has been known to predict accurate protein secondary structures as high as 92% (Wishart & Sykes. 1994).
Chemical shift outliers and reference correction
 Chemical shift reference correction and chemical shift outliers are detected by LACS algorithm (Wang et al. 2005). Probabilistic text file (temp_LACS.out) and NDP-PLOT files (ndpplot_LACS_CACB_CA.ini and etc) are provided. From LACS results, X axis refers differences between secondary chemical shifts of CA and CB, while Y axis refers secondary chemical shifts of target atom (CA, CB, CO, etc). Residues far from a fitted line are flagged.
Chemical shift reference correction and chemical shift outliers are detected by LACS algorithm (Wang et al. 2005). Probabilistic text file (temp_LACS.out) and NDP-PLOT files (ndpplot_LACS_CACB_CA.ini and etc) are provided. From LACS results, X axis refers differences between secondary chemical shifts of CA and CB, while Y axis refers secondary chemical shifts of target atom (CA, CB, CO, etc). Residues far from a fitted line are flagged.
Hydrophobicity
 Exposure to water for each residue is predicted by searching PACSY database with assigned backbone chemical shifts on-the-fly. I-PINE uses Buried (0%-10%; red), Medium (10%-30%; purple), Exposed (30%-100%; blue) criteria to categorize by using SASA (Solvent Accessible Surface Area; Lee & Richards 1971). The SASA of Gly-X-Gly is 100% exposure to water, and the categorization is relative to that. Statistical counting of tripeptide in PACSY with backbone chemical shifts with 1.0 ppm tolerance is conducted. A text report (core_report.txt) and a NDPPLOT file (ndpplot_CORE.ini) are provided. Also provided is pacsy_entries.txt in case proteins with same sequences exist in the PDBSEQ_DB table of PACSY database.
Exposure to water for each residue is predicted by searching PACSY database with assigned backbone chemical shifts on-the-fly. I-PINE uses Buried (0%-10%; red), Medium (10%-30%; purple), Exposed (30%-100%; blue) criteria to categorize by using SASA (Solvent Accessible Surface Area; Lee & Richards 1971). The SASA of Gly-X-Gly is 100% exposure to water, and the categorization is relative to that. Statistical counting of tripeptide in PACSY with backbone chemical shifts with 1.0 ppm tolerance is conducted. A text report (core_report.txt) and a NDPPLOT file (ndpplot_CORE.ini) are provided. Also provided is pacsy_entries.txt in case proteins with same sequences exist in the PDBSEQ_DB table of PACSY database.
TALOS-N
 TALOS-N is an aritificial neural network (ANN) based hybrid system for empirical prediction of protein backbone φ/ψ torsion angles, sidechain χ1 torsion angles and secondary structure using a combination of six kinds (HN, Hα, Cα, Cβ, CO, N) of chemical shift assignments for given residue sequence (Shen and Bax 2013). I-PINE runs TALOS-N with chemical shifts assigned by its algorithm subsequently, and also generates torsion angle restraint files can be readily used by other structure calculation programs like CYANA, XPLOR-NIH and PONDEROSA-C/S. It also generates RCI-S2 (Random Coil Index Order Parameter; Berjanskii and Wishart 2005) that predicts protein motions. NDPPLOT files for secondary structures (ndpplot_talosn_ss.ini) and RCI-S2 (ndpplot_talosn_s2.ini) are provided.
TALOS-N is an aritificial neural network (ANN) based hybrid system for empirical prediction of protein backbone φ/ψ torsion angles, sidechain χ1 torsion angles and secondary structure using a combination of six kinds (HN, Hα, Cα, Cβ, CO, N) of chemical shift assignments for given residue sequence (Shen and Bax 2013). I-PINE runs TALOS-N with chemical shifts assigned by its algorithm subsequently, and also generates torsion angle restraint files can be readily used by other structure calculation programs like CYANA, XPLOR-NIH and PONDEROSA-C/S. It also generates RCI-S2 (Random Coil Index Order Parameter; Berjanskii and Wishart 2005) that predicts protein motions. NDPPLOT files for secondary structures (ndpplot_talosn_ss.ini) and RCI-S2 (ndpplot_talosn_s2.ini) are provided.
CS-Rosetta (optional)
 If a user enables "Run BMRB CS-Rosetta" option, I-PINE submits 3D structure calculation jobs to the CS-Rosetta web server hosted by BMRB. This takes signifantly longer than chemical shift assignments, and the time durates depending on the size of the protein and complexity of the fold. Therefore, I-PINE sends other results to the user first, and starts CS-Rosetta next. You will receive emails from BMRB CS-Rosetta web server notifying progresses and an email from I-PINE that your results URL also includes the 3D structures when the calculation finishes. Also, the results will be automatically imported to I-PINE results with the job identification nubmber.
If a user enables "Run BMRB CS-Rosetta" option, I-PINE submits 3D structure calculation jobs to the CS-Rosetta web server hosted by BMRB. This takes signifantly longer than chemical shift assignments, and the time durates depending on the size of the protein and complexity of the fold. Therefore, I-PINE sends other results to the user first, and starts CS-Rosetta next. You will receive emails from BMRB CS-Rosetta web server notifying progresses and an email from I-PINE that your results URL also includes the 3D structures when the calculation finishes. Also, the results will be automatically imported to I-PINE results with the job identification nubmber.
PONDEROSA (optional)
 If a user submits a job with 3D NOESY-CHSQC and 3D NOESY-NHSQC together, I-PINE estimates completeness level of resonance assignments and NOE cross peaks in the NOESY data if it will be worthy to proceed an automated 3D structure calculation using PONDEROSA web server hosted by NMRFAM. The criteria to run is completeness 80% of resonances achieving 0.7 or higher RCI-S2 values. Tolerances are set to 0.4 ppm, 0.4 ppm and 0.03 ppm for nitrogen, carbon and proton respectively. A user will receive notification emails from ARECA and PONDEROSA web server when an email address is provided (optional). A user can use full features of PONDEROSA Analyzer program to analyzer, validate results and resubmit with refined constraints.
If a user submits a job with 3D NOESY-CHSQC and 3D NOESY-NHSQC together, I-PINE estimates completeness level of resonance assignments and NOE cross peaks in the NOESY data if it will be worthy to proceed an automated 3D structure calculation using PONDEROSA web server hosted by NMRFAM. The criteria to run is completeness 80% of resonances achieving 0.7 or higher RCI-S2 values. Tolerances are set to 0.4 ppm, 0.4 ppm and 0.03 ppm for nitrogen, carbon and proton respectively. A user will receive notification emails from ARECA and PONDEROSA web server when an email address is provided (optional). A user can use full features of PONDEROSA Analyzer program to analyzer, validate results and resubmit with refined constraints.
Cysteine oxidation state
 If there are cysteines in sequences, I-PINE assigns their ionization states if they are oxidized or reduced using assigned CB chemical shifts. It has been known that CB chemical shifts represent redox states of a cysteine (background), and we calculate secondary chemical shifts between observed CB and random coil CB of reduced/oxidized state (background) and a provide predicted state with probability using a gaussian function.
If there are cysteines in sequences, I-PINE assigns their ionization states if they are oxidized or reduced using assigned CB chemical shifts. It has been known that CB chemical shifts represent redox states of a cysteine (background), and we calculate secondary chemical shifts between observed CB and random coil CB of reduced/oxidized state (background) and a provide predicted state with probability using a gaussian function.
Proline isomerization state
 I-PINE assigns prolines' isomerization states if they are trans- or cis- using assigned C, CB and CG chemical shifts. It uses histograms made by Yang Shen from NIH (PROMEGA) and a provide predicted state with probability using histogram-derived probability density functions.
I-PINE assigns prolines' isomerization states if they are trans- or cis- using assigned C, CB and CG chemical shifts. It uses histograms made by Yang Shen from NIH (PROMEGA) and a provide predicted state with probability using histogram-derived probability density functions.
PyMOL scripts
PyMOL scripts to visualize various information are provided; assignment probabilities (pine_pymol), propensity to be buried in the core (core_pymol), and flexibilities (rci_pymol). Each script file can be used while 3D structure is loaded by typing @script (e.g. @pine_pymol).
 I-PINE submission form is a web-based user interface (WUI) to submit a job. It stays on the right in any of the I-PINE web pages (just righthand side of this text). A user can simply select input files by clicking "Choose File" button in the web interface or typing user information. When all the inputs are set and ready to submit a job, a user can click "Submit" button at the bottom in the I-PINE submission form. Job ID and a link to the web report page (example) will appear.
I-PINE submission form is a web-based user interface (WUI) to submit a job. It stays on the right in any of the I-PINE web pages (just righthand side of this text). A user can simply select input files by clicking "Choose File" button in the web interface or typing user information. When all the inputs are set and ready to submit a job, a user can click "Submit" button at the bottom in the I-PINE submission form. Job ID and a link to the web report page (example) will appear.
 PINE-SPARKY.2 is a graphical user interface (GUI) that enables user to use I-PINE in the NMRFAM-SPARKY package. It comes as a pre-installed plug-in extension (two-letter-code ep) to the NMRFAM-SPARKY. It is highly recommended to use this program to reduce the input file preparation time preparing and avoid formatting errors.
PINE-SPARKY.2 is a graphical user interface (GUI) that enables user to use I-PINE in the NMRFAM-SPARKY package. It comes as a pre-installed plug-in extension (two-letter-code ep) to the NMRFAM-SPARKY. It is highly recommended to use this program to reduce the input file preparation time preparing and avoid formatting errors.
A simple python script, pyIPINE.py, is developed to provide a command-line user interface (CLI) to use I-PINE in the terminal by users or by wrapper scripts or other programs. Help for pyIPINE.py can be displayed by $ python pyIPINE.py -h while an example workflow can be displayed by $ python pyIPINE.py -e.
We made I-PINE/ssPINE user group recently. Please post a question in the user group or find a solution there, or ask NMRFAM directly. We encourage your participation in the I-PINE/ssPINE user group while it can help others facing similar problems.
We have tested a variety of operating systems and web browsers other than listed here. Please let use know if you have any issues.
| OS | Version | Chrome | Firefox | Microsoft Edge | Safari |
| Linux | Linux Mint 18.3 | 70.0.3538.110 | 63.03 | n/a | n/a |
| MacOS | Sierra | 71.0.3578.98 | 56.0.2 | n/a | 7.0.2 |
| Windows | 10 | 71.0.3578.80 | 64.0 | 44.17763.1.0 | n/a |